随着基因治疗领域的快速发展,AAV载体因其较高的转染效率和安全性成为了一种最常用的基因治疗病毒载体。然而,在其CMC开发过程中仍面临许多挑战,例如复杂的生产工艺、产品质量的一致性以及规模化生产的稳定性等问题。类似于生物大分子药物(例如单抗),FDA也希望基因治疗产品能够遵循QbD理念,使得产品开发过程更加具有系统性、可控性和可预测性,实现生产工艺的稳健和产品质量的稳定可靠。
本文将简要介绍QbD基本原则及其在AAV产品CMC开发中的应用,说明QbD方法对科学、高效开展基因治疗产品CMC开发的作用。
- 确定产品目标质量概况(Quality Target Product Profile, QTPP)首先定义AAV基因治疗产品的QTPP,包括疗效、安全性、稳定性等关键质量属性(Critical Quality Attributes, CQAs),这是建立产品质量标准和开发过程的基础。
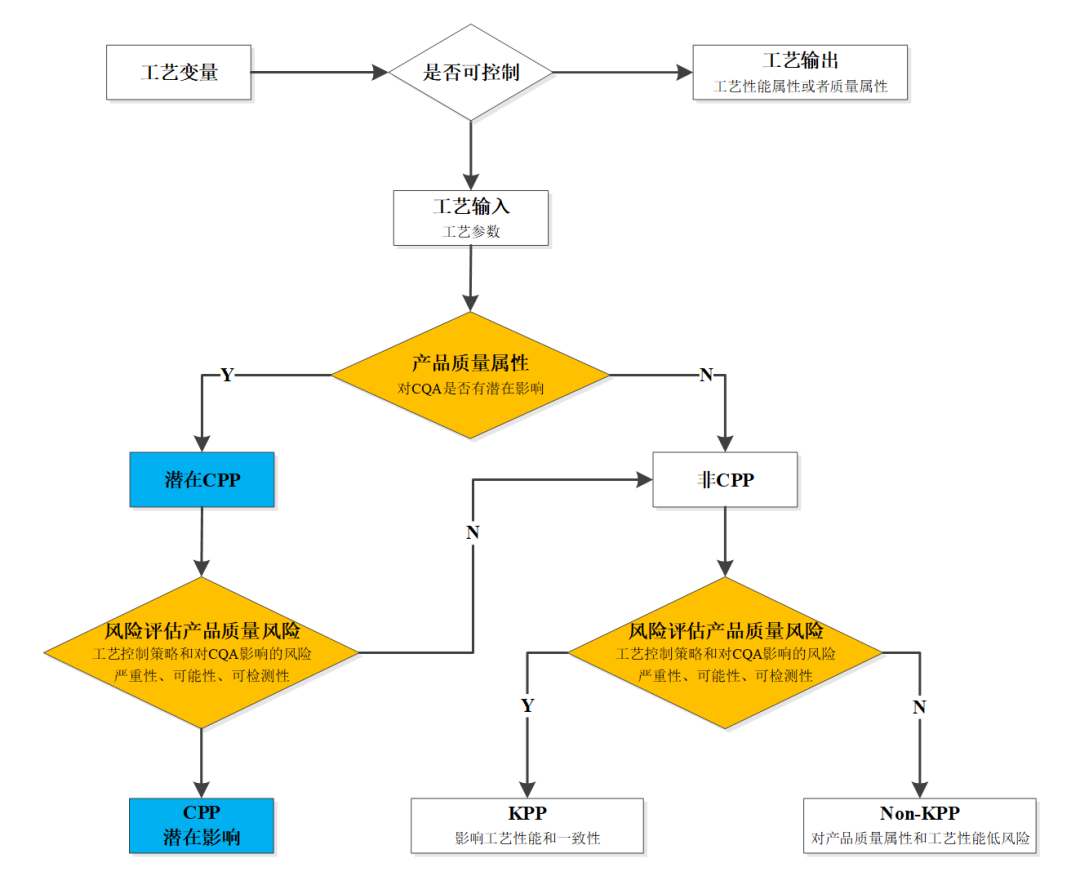
- 识别关键质量属性(CQAs)通过风险评估方法识别影响AAV疗效和安全性的CQAs,如基因组滴度、感染滴度、效力、纯度等,每个CQA都应适当控制以保证产品质量。
- 确定关键工艺参数(Critical Process Parameters, CPPs)和关键物料属性(Critical Material Attributes, CMAs)通过实验确定影响CQAs的CPPs和CMAs,例如细胞培养条件、病毒收获条件、纯化步骤参数等,对这些参数进行优化,以确保生产工艺的可重复性和产品质量的一致性。
- 制定风险管理计划采用故障模式与效应分析(Failure Mode and Effects Analysis, FMEA)等工具来评估CQAs受CPPs和CMAs影响的潜在风险,制定相应的风险缓解策略和监控计划。
- 开发设计空间(Design Space)在确定了CPPs和CMAs对CQAs影响的基础上,开发设计空间(即参数的范围),只要在这个范围内操作,就能够保证产品CQAs的一致性。
- 确立控制策略基于以上步骤,制定详细的控制策略,以确保在整个生产过程中CQAs处于可接受的范围之内。
典型的与基因治疗产品开发阶段相适应的CMC开发策略如图-1所示:
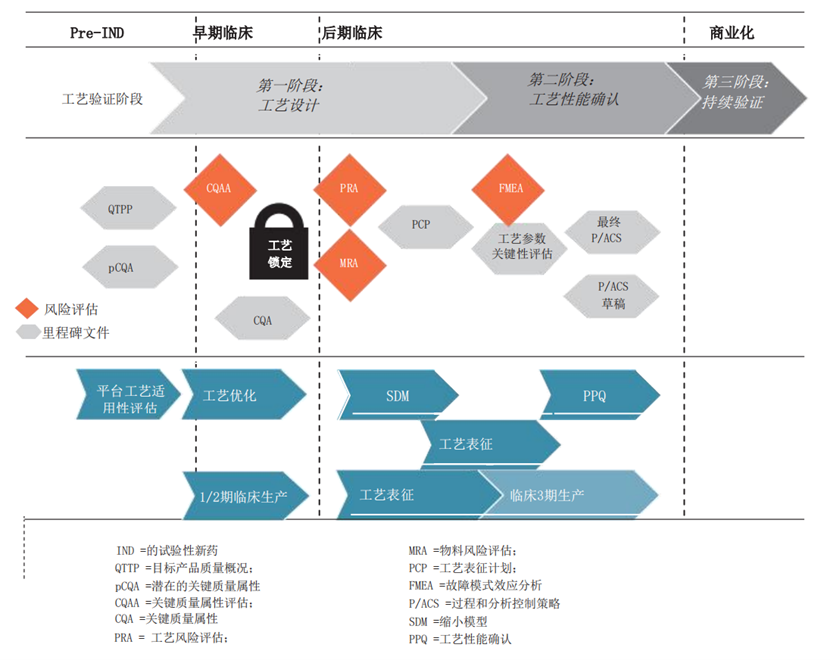
图-1:与产品开发阶段相适应的CMC开发策略
许多基因治疗产品针对的是那些临床急需,但尚未得到满足的治疗需求,因此该类产品极可能适合申请快速审批途径,例如突破性疗法、快速通道、优先审评和加速审评。虽然可允许基于II期临床试验数据或单中心III期临床试验数据提交BLA上市申请,但是仍然需要完整的CMC资料作为支撑,包括工艺开发和验证。因此,如果某个产品希望通过国内外审评机构的快速审评通道上市,应在早期工艺开发阶段就引入QbD方法,以满足紧凑的审批时限要求。